Overview
The extra-cellular RNA processing toolkit (exceRpt) was designed to handle the variable contamination and often poor quality data obtained from low input smallRNA-seq samples such as those obtained from extra-cellular preparations. However the tool is perfectly capable of processing data from more standard cellular preparations and, with minor modifications to the command-line call, is also capable of processing WGS/exome and long RNA-seq data.
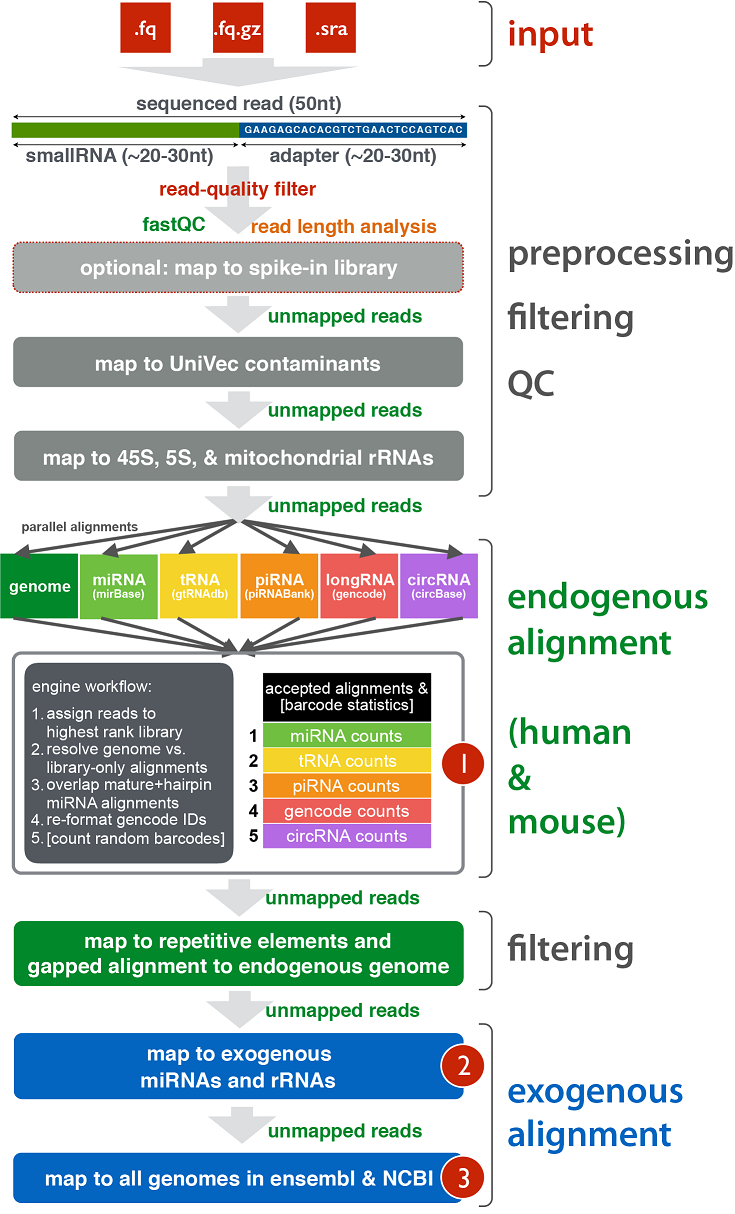
The schematic diagram below details the various stages of the exceRpt pipeline including pre-processing of the raw reads, contaminant removal, alignment to the endogenous (human/mouse) genome and annotated transcriptome, and finally alignment to a large number of exogenous sequences for detection of foreign nucleic acids.
How to use exceRpt
Genboree.org
Signing up for an account on Genboree.org is by far the fastest way to start using exceRpt. The Genboree Workbench provides a graphical interface to exceRpt that is free for academic use. Set up a free account to use the Genboree Workbench, upload some data, and start using exceRpt, all via the browser (instructions available here).
Docker
Slightly more effort to get set up than using Genboree, but using the Docker image is the best way to use exceRpt on your own machine/cluster. The exceRpt Docker image is pre-baked with all required dependencies and should ‘just work’ with minimal installation. Docker is native on Red Hat Enterprise Linux distributions (RHEL) and also available on Amazon, enabling exceRpt to be run in the cloud. Instructions are available for installing Docker on Mac OS X, Linux, Windows, and Amazon Web Services.
BEWARE, exceRpt requires about 16GB of RAM in order to function properly, runs on machines with less than this amount of available memory will very likely fail. If running Docker inside a virtual machine (such as via VirtualBox on Mac OS X) it is important to check that the VM has been initialised with at least 16GB of memory. This is usually a setting that can be modified as long as the VM is not currently running. RNA-seq read pre-processing can be achieved using much less memory, however much of the useful output of the tool (e.g. all alignments / transcript quantifications) will be unavailable.
Machines with sufficient memory will almost certainly be equipped with a sufficient CPU, however machines with fewer than 4 CPU cores may struggle to run the alignments required by exceRpt.
As with the manual installation described below, you will also need to download the pre-compiled genome and transcriptome indices for exceRpt to use. These are too large to be included in the Docker image (each uncompressed reference index requires around 35GB on disk), but using these links you can download the exceRpt database files:
There are two other optional exceRpt databases for completing the analysis of endogenous-unmapped reads by mapping to all known exogenous miRNAs, rRNAs, and genomes. The miRNA sequences are obtained from miRBase, ribosomal-RNA sequences from the Ribosome Database Project, and the full-genomes of all sequenced bacteria, viruses, funghi, plants, protists, and ‘edible’ eukaryotes obtained from Ensembl and the NCBI. These two exogenous databases are available at the locations below (beware the very large size of the exogenous genomes database, which is around 1.5 TB and will take a long time to download). When running the Docker image, including these exogenous databases requires a slight modification to the standard ‘docker run’ command - please contact the exceRpt authors for further information.
Once you have downloaded the desired genome database and have successfully installed and started Docker on your machine/cluster/AWS-instance only a few commands are required to install and run exceRpt.
Install exceRpt
To download the exceRpt Docker image, type:
docker pull rkitchen/excerpt
Download the database
To download the exceRpt database for your genome/species of interest (currently one of hg19, hg39, or mm10) use the following commands. Here we’ll download the latest build of the human genome and transcriptome (hg38):
mkdir ~/DirectoryContainingMyexceRptDatabase
cd ~/DirectoryContainingMyexceRptDatabase
wget http://org.gersteinlab.excerpt.s3-website-us-east-1.amazonaws.com/exceRptDB_v4_hg38_lowmem.tgz
tar -xvf exceRptDB_v4_hg38_lowmem.tgz
Grab some test smRNA-seq data
To run a smallRNA-seq sample through the exceRpt pipeline we can grab a publicly-available dataset from the SRA:
mkdir ~/DirectoryContainingMyInputSample
cd ~/DirectoryContainingMyInputSample
wget ftp://ftp.ddbj.nig.ac.jp/ddbj_database/dra/sralite/ByExp/litesra/SRX/SRX010/SRX010851/SRR026761/SRR026761.sra
Alternatively, the exceRpt docker image comes pre-loaded with a tiny (1,000 read) human smRNA-seq dataset. This can be run without the need to download any data, you’ll just need to specify a database and output location and use the following input file:
INPUT_FILE_PATH=/exceRptInput/testData_human.fastq.gz
Specify an output directory
Almost done, just need to make sure there is a directory in which to put the exceRpt pipeline output:
mkdir ~/DirectoryInWhichToPutMyResults
Run exceRpt
Finally, we can input this sample to the exceRpt pipeline, aligning to the hg38 genome+transcriptome we just downloaded:
docker run -v ~/DirectoryContainingMyInputSample:/exceRptInput \
-v ~/DirectoryInWhichToPutMyResults:/exceRptOutput \
-v ~/DirectoryContainingMyexceRptDatabase/hg38:/exceRpt_DB/hg38 \
-t rkitchen/excerpt \
INPUT_FILE_PATH=/exceRptInput/SRR026761.sra
Most of the Docker command is loading directories on your machine (the -v parameters) so that exceRpt can read from or write to them. The directory to the left of each : can obviously be whatever you want, but it is important to make sure the right side of each : is written as above or exceRpt will not be able to find/write the data it needs.
Again, if you’re wanting to run the built-in 1,000 read human smRNA-seq example dataset, the command to run would be:
docker run -v ~/DirectoryInWhichToPutMyResults:/exceRptOutput \
-v ~/DirectoryContainingMyexceRptDatabase/hg38:/exceRpt_DB/hg38 \
-t rkitchen/excerpt \
INPUT_FILE_PATH=/exceRptInput/testData_human.fastq.gz
Passing arguments to exceRpt
The last line of the docker run command (INPUT_FILE_PATH=) is a direct call to exceRpt itself. As such, any customisable options should be specified this way. For example we could re-order the priority of the transcriptome libraries (for example if we expected a sample to have more long-RNA fragments than miRNAs) using the following call to docker:
docker run -v ~/DirectoryContainingMyInputSample:/exceRptInput \
-v ~/DirectoryInWhichToPutMyResults:/exceRptOutput \
-v ~/DirectoryContainingMyexceRptDatabase/hg38:/exceRpt_DB/hg38 \
-t rkitchen/excerpt \
INPUT_FILE_PATH=/exceRptInput/SRR026761.sra \
ENDOGENOUS_LIB_PRIORITY=gencode,miRNA,tRNA,piRNA,circRNA
We can also use this approach to change the genome to which we are aligning (for example, change to mouse mm10)
docker run -v ~/DirectoryContainingMyInputSample:/exceRptInput \
-v ~/DirectoryInWhichToPutMyResults:/exceRptOutput \
-v ~/DirectoryContainingMyexceRptDatabase/mm10:/exceRpt_DB/mm10 \
-t rkitchen/excerpt \
INPUT_FILE_PATH=/exceRptInput/SRR026761.sra \
MAIN_ORGANISM_GENOME_ID=mm10
Finally, if you’re lucky enough to have an abundance of available disk space, it is easier in the long run to pass Docker the directory containing all available exceRpt genome databases so that to change organism/genome only the MAIN_ORGANISM_GENOME_ID variable needs to be modified:
docker run -v ~/DirectoryContainingMyInputSample:/exceRptInput \
-v ~/DirectoryInWhichToPutMyResults:/exceRptOutput \
-v ~/DirectoryContainingMyexceRptDatabase:/exceRpt_DB \
-t rkitchen/excerpt \
INPUT_FILE_PATH=/exceRptInput/SRR026761.sra \
MAIN_ORGANISM_GENOME_ID=mm10 \
[OPTION]=[VALUE]
Analysis customisation options
There are a number of options available for customising the analysis that are specified using the command-line. These are a list of the most commonly-modified options and their default values.
Required OPTIONs:
INPUT_FILE_PATH | Path to the input fastq/fasta/sra file
Main analysis OPTIONs:
ADAPTER_SEQ | 'guessKnown'/'none'/<String> | [default: 'guessKnown'] will attempt to guess the 3 adapter using known sequences. The actual adapter can be input here if known, or specify 'none' if the adapter is already removed
SAMPLE_NAME | <String> | add an optional ID to the input file specified above
MAIN_ORGANISM_GENOME_ID | 'hg38'/'hg19'/'mm10' | [default: 'hg38'] changes the organism/genome build used for alignment
CALIBRATOR_LIBRARY | <Path> | path to a bowtie2 index of calibrator oligos used for QC or normalisation
ENDOGENOUS_LIB_PRIORITY | <comma,separated,list,no,spaces> | [default: 'miRNA,tRNA,piRNA,gencode,circRNA'] choose the priority of each library during read assignment and quantification
Additional analysis OPTIONs:
TRIM_N_BASES_5p | <int> | [default: '0'] remove N bases from the 5' end of every read
TRIM_N_BASES_3p | <int> | [default: '0'] remove N bases from the 3' end of every read
RANDOM_BARCODE_LENGTH | <int> | [default: 0] identify and remove random barcodes of this number of nucleotides. For a Bioo prep with a 4N random barcode on both the 3' and 5' adapter, this value should be '4'.
RANDOM_BARCODE_LOCATION | '-5p -3p'/'-5p'/'-3p' | [default: '-5p -3p'] specify where to look for the random barcode(s)
KEEP_RANDOM_BARCODE_STATS | 'false'/'true' | [default: 'false'] specify whether or not to calculate overrepresentation statistics using the random barcodes (this may be slow and memory intensive!)
DOWNSAMPLE_RNA_READS | <int> | [default: NULL] choose whether to downsample to this number of reads after assigning reads to the various transcriptome libraries (may be useful for normalising very different yields)
Hardware-specific OPTIONs:
N_THREADS | <int> | [default: 4] change the number of threads used in the alignments performed by exceRpt
JAVA_RAM | <String> | [default: '10G'] change the amount of memory (RAM) available to Java. This may need to be higher if crashes occur during quantification or random barcode stats calculation
REMOVE_LARGE_INTERMEDIATE_FILES | 'false'/'true' | [default: 'false'] when exceRpt finishes, choose whether to remove the large alignment files that can take a lot of disk space
Alignment/QC OPTIONs:
MIN_READ_LENGTH | <int> | [default: 18] minimum read-length to use after adapter (+ random barcode) removal
QFILTER_MIN_QUAL | <int> | [default: 20] minimum base-call quality of the read
QFILTER_MIN_READ_FRAC | <double> | [default: 80] read must have base-calls higher than the value above for at least this fraction of its length
STAR_alignEndsType | 'Local'/'EndToEnd' | [default: Local] defines the alignment mode; local alignment is recommended to allow for isomiRs
STAR_outFilterMatchNmin | <int> | [default: 18] minimum number of bases to include in the alignment (should match the minimum read length defined above)
STAR_outFilterMatchNminOverLread | <double> | [default: 0.9] minimum fraction of the read that *must* remain following soft-clipping in a local alignment
STAR_outFilterMismatchNmax | <int> | [default: 1] maximum allowed mismatched bases in the aligned portion of the read
MAX_MISMATCHES_EXOGENOUS | <int> | [default: 0] maximum allowed mismatched bases in the *entire* read when aligning to exogenous sequences
Manual installation
This is the most labour-intensive mode of using exceRpt, but it offers the most flexibility. Generally not recommended unless you are very familiar with the *NIX environment, shell scripting, and makefile syntax. Detailed instructions on how to install exceRpt and its various dependencies will [one day] be listed toward the bottom of this page.
In addition to the genome+transcriptome indices listed in the Docker section above, you will also require the core database that is contained within the Docker image. The CORE database can be downloaded here. Once this is extracted, it will create a DATABASE directory, into which the genome+transcriptome databases can be extracted. If the DATABASE directory is located at the same path as the exceRpt_smallRNA makefile, it will be automatically detected. If it is preferable to keep the DATABASE in a different location to the rest of the exceRpt executables and dependencies, this new path can be specified at runtime using the make flag: DATABASE_PATH=/path/to/exceRpt/DATABASE
Once all dependencies are installed, the exceRpt pipeline can be run on a human sample (here an SRA binary file) using default parameters using the following skeleton command:
make -f exceRpt_smallRNA \
INPUT_FILE_PATH=~/DirectoryContainingMyInputSample/SRR026761.sra \
OUTPUT_DIR=~/DirectoryInWhichToPutMyResults
It is important to properly configure the makefile for this to work. It is probably a good idea to get in touch with the maintainers using the email address listed at the bottom of the page before embarking on this yourself!
Understanding the exceRpt output contained in OUTPUT_DIR
A variety of output files are created for each sample as they are run through the exceRpt pipeline. At the highest level, 5 files and one directory are output to the OUTPUT_DIR:
[sampleID]/ | Directory containing the complete set of output files for this sample
[sampleID]_CORE_RESULTS_v*.tgz | Archive containing only the most commonly used results for this sample
[sampleID].err | Text file containing error logging information for this run
[sampleID].log | Text file containing normal logging information for this run
[sampleID].qcResult | Text file containing a variety of QC metrics for this sample
[sampleID].stats | Text file containing a variety of alignment statistics for this sample
[sampleID]_CORE_RESULTS_v4.*.tgz
This archive contains the most commonly used results for this sample and is the only file required to run the mergePipelineRuns.R script described below for processing the output from multiple runs of the exceRpt pipeline (i.e. for multiple samples). The contents of this archive are as follows:
[sampleID].log | Same as above
[sampleID].stats | Same as above
[sampleID].qcResult | Same as above
[sampleID]/[sampleID].readCounts_*_sense.txt | Read counts of each annotated RNA using sense alignments
[sampleID]/[sampleID].readCounts_*_sense.txt | Read counts of each annotated RNA using antisense alignments
[sampleID]/[sampleID].*.coverage.txt | Contains read-depth across all gencode transcripts
[sampleID]/[sampleID].*.CIGARstats.txt | Summary of the alignment characteristics for genome-mapped reads
[sampleID]/[sampleID].*_fastqc.zip | FastQC output both before and after UniVec/rRNA contaminant removal
[sampleID]/[sampleID].*.readLengths.txt | Counts of the number of reads of each length following adapter removal
[sampleID]/[sampleID].*.counts | Read counts mapped to UniVec & rRNA (and calibrator oligo, if used) sequences
[sampleID]/[sampleID].*.knownAdapterSeq | 3' adapter sequence guessed (from known adapters) in this sample
[sampleID]/[sampleID].*.adapterSeq | 3' adapter used to clip the reads in this run
[sampleID]/[sampleID].*.qualityEncoding | PHRED encoding guessed for the input sequence reads
[sampleID]/
The main results directory contains all files above as well as the following:
Intermediate files containing reads ‘surviving’ each stage, in the following order of 1) 3’ adapter clipping, 2) 5’/3’ end trimming, 3) read-quality and homopolymer filtering, 4) UniVec contaminant removal, and 5) rRNA removal:
[sampleID]/[sampleID].*.fastq.gz | Reads remaining after each QC / filtering / alignment step
Reads aligned at each step of the pipeline in the following order 1) UniVec, 2) rRNA, 3) endogenous genome, 4) endogenous transcriptome:
[sampleID]/filteringAlignments_*.bam | Alignments to the UniVec and rRNA sequences
[sampleID]/endogenousAlignments_genome*.bam | Alignments (ungapped) to the endogenous genome
[sampleID]/endogenousAlignments_genomeMapped_transcriptome*.bam | Transcriptome alignments (ungapped) of reads mapped to the genome
[sampleID]/endogenousAlignments_genomeUnmapped_transcriptome*.bam | Transcriptome alignments (ungapped) of reads **not** mapped to the genome
Alignment summary information obtained after invoking the library priority. In the default setting, this will choose a miRBase alignment over any other alignment, for example if it is aligned to both a miRNA in miRBase and a miRNA in Gencode, the miRBase alignment is kept and all others discarded. It is especially important for tRNAs to be chosen in favour of piRNAs, as the latter have quite a large number of mis-annotations to the former.
[sampleID]/endogenousAlignments_Accepted.txt.gz | All compatible alignments against the transcriptome after invoking the library priority
[sampleID]/endogenousAlignments_Accepted.dict | Contains the ID(s) of the RNA annotations indexed in the fifth column of the .txt.gz file above
Finally, the quantifications are stored in the various readCounts_*.txt files. The format of these tab-delimited files is as follows:
ReferenceID uniqueReadCount totalReadCount multimapAdjustedReadCount multimapAdjustedBarcodeCount
hsa-miR-143-3p:MIMAT0000435:Homo:sapiens:miR-143-3p 1235 4147219 4147219.0 0.0
hsa-miR-10b-5p:MIMAT0000254:Homo:sapiens:miR-10b-5p 1430 2420500 2420241.0 0.0
hsa-miR-10a-5p:MIMAT0000253:Homo:sapiens:miR-10a-5p 1115 784863 784600.5 0.0
hsa-miR-192-5p:MIMAT0000222:Homo:sapiens:miR-192-5p 759 559068 558542.5 0.0
Where ReferenceID is the ID of this annotated RNA, uniqueReadCount is the number of unique insert sequences attributed to this annotated RNA, totalReadCount is the total number of reads attributable to this annotated RNA, multimapAdjustedReadCount is the count after adjusting for multi-mapped reads, and multimapAdjustedBarcodeCount (available only for samples prepped with randomly barcoded 5’ and/or 3’ adapters such as Bioo) is the number of unique N-mer barcodes adjusted for multimapping ambiguity in the insert sequence.
Processing exceRpt output from multiple samples
Also provided is a script to combine output from multiple samples run through the exceRpt pipeline. The script (mergePipelineRuns.R) will take as input a directory containing 1 or more subdirectories or zipfiles containing output from the makefile above. In this way, results from 1 or more smallRNA-seq samples can be combined, several QC plots are generated, and the read-counts are normalised ready for downstream analysis by clustering and/or differential expression.
Installation
This script is comparatively much simpler to install. Once the R software (http://cran.r-project.org/) is set up on your system the script should automatically identify and install all required dependencies. Again, this script is available on the Genboree Workbench (www.genboree.org) and is also free for academic use.
Using the script
On the command line
The Rscript application provides a neat means of running the script:
Rscript mergePipelineRuns.R ~/DirectoryContainingMyexceRptRuns
Here, the directory in which the script will search for valid exceRpt output (~/DirectoryContainingMyexceRptRuns) should be the same as the (~/DirectoryInWhichToPutMyResults) specific when running each sample through exceRpt.
Interactively in R
Alternatively in an interactive R session, the merge can be performed using the following two commands:
> source("mergePipelineRuns_functions.R")
> processSamplesInDir(data.dir, output.dir)
Apart from some status messages, warnings, or possibly errors, no R objects are output from this function. Instead several files are created that are described immediately below…
Script output
Several files are output by the script in the location of the input exceRpt results (or somewhere else if explicitly specified). All output files are prefixed with ‘exceRpt_’ and contain a variety of information regarding all samples input:
| File Name | Description |
|---|---|
| QC data: | |
exceRpt_DiagnosticPlots.pdf |
All diagnostic plots automatically generated by the merge script |
exceRpt_readMappingSummary.txt |
Read-alignment summary including total counts for each library |
exceRpt_ReadLengths.txt |
Read-lengths (after 3’ adapters/barcodes are removed) |
| Raw transcriptome quantifications: | |
exceRpt_miRNA_ReadCounts.txt |
miRNA read-counts quantifications |
exceRpt_tRNA_ReadCounts.txt |
tRNA read-counts quantifications |
exceRpt_piRNA_ReadCounts.txt |
piRNA read-counts quantifications |
exceRpt_gencode_ReadCounts.txt |
gencode read-counts quantifications |
exceRpt_circularRNA_ReadCounts.txt |
circularRNA read-count quantifications |
| Normalised transcriptome quantifications: | |
exceRpt_miRNA_ReadsPerMillion.txt |
miRNA RPM quantifications |
exceRpt_tRNA_ReadsPerMillion.txt |
tRNA RPM quantifications |
exceRpt_piRNA_ReadsPerMillion.txt |
piRNA RPM quantifications |
exceRpt_gencode_ReadsPerMillion.txt |
gencode RPM quantifications |
exceRpt_circularRNA_ReadsPerMillion.txt |
circularRNA RPM quantifications |
| R objects: | |
exceRpt_smallRNAQuants_ReadCounts.RData |
All raw data (binary R object) |
exceRpt_smallRNAQuants_ReadsPerMillion.RData |
All normalised data (binary R object) |
Manual Installation
Dependencies
Core dependencies
- Java v1.8+
- fastx v0.0.14+
- STAR v2.4.2a+ - required for most exogenous alignments
- bowtie2 v2.2.4+
- samtools v1.3+
- fastQC v0.11.7+
Optional dependencies
- sratoolkit v2.5.1+ - required for processing .sra files from NCBI archival data
Installation steps (incomplete)
- Install dependencies
- Download exceRpt database(s)
-
Configure the makefile, especially the following variable:
EXE_DIR := [location of the pipeline, database, and dependencies]
Support
Rob Kitchen; r [dot] r [dot] kitchen [at] gmail [dot] com